
基于HMM的基因识别并行计算
1 引言
本文引用地址:http://www.amcfsurvey.com/article/198952.htm20世纪90年代以来,伴随着各种基因组测序计划的展开和分子结构测定技术的突破,数以百计的生物学数据库如雨后春笋般迅速出现和成长。如何利用这些不断爆炸性增长的有关生物分子的原始数据,有效解决基因识别问题显得越来越迫切。最初的基因分析方法是进行简单的核苷酸统计,而后加上剪切保守位点的检测。以后采用了人工神经网络、隐马尔科夫模型(HMM)[1,2]等先进的信息处理和分析技术,提高基因识别的准确率。但由于生物信息数据量巨大,传统的串行算法往往无法处理或难以在满意的时间内得到结果。本文针对基因序列的识别,讨论隐马尔科夫模型分析算法的并行算法设计和并行效果分析。
2 隐马尔科夫模型法
隐马尔科夫模型[3](Hidden Markov Models,HMM)是一种概率论模型,这种方法已经成功应用于多个领域,如语音识别、光学字符识别等。HMM在生物信息学领域中也有着重要的应用,如序列分析、基因识别等。目前,基因识别的HMM方法也大致可以分为两类,一类为按照内容搜索的方法,通过核苷酸和三联密码子等在编码区的分布规律来界定蛋白质的编码区;另一类为按照信号搜索的方法,通过编码区周围的信号界定蛋白质编码区。
2.1 马尔科夫链
考虑只取有限个或可数个状态的随机过程{Xn,n=0,1,2,…},假设对一切状态i0,i1,…,in-1,i,j和一切n≥0,有P{Xn+1=j | Xn=i,Xn-1=in-1,…,X1=i1,X0=i0} = P{Xn+1=j | Xn=i}成立,则称此随机过程为离散状态马尔科夫链。简单的说,就是系统未来的状态仅依赖于当前状态。一个马尔科夫链的概率分布完全由它的初始分布P(X0)与转移矩阵P=(pij)决定。
2.2 HMM基本原理
隐马尔科夫模型HMM是由马尔科夫链发展扩充而来的一种随机模型。HMM可以被理解为一个双重随机过程,一个是不可观察的(隐含的)状态变化序列,另一个是由该不可观察的状态产生的可观察符号序列。隐马尔科夫模型形式描述如下:一个HMM模型是一个三元组M=(A,S,Q),其中A是字母表,S是有限状态集合,每个状态可以释放字母表中的字符。Q为概率集合,包括两个部分:一是状态转换概率fkl,k,l∈S,表示从状态k转化到状态l的概率;二是字符释放概率,记为ek(b) (k∈S,b∈A),表示在状态k下释放出字符b的概率。令路径Π=(π1,π2,…,πL )是模型M的一个相继状态序列,X=(x1,x2,…,xL)是一个字符序列,按下述方式定义状态转换概率和字符释放概率:
fkl = p(πi = l|πi-1 = k)
ek(b) = p(xi=b|πi= k)
对于给定的路径Π,可以按下面的公式计算出产生序列X的概率:
P(X|Π)= fπ0,π1 eπi (xi)fπi,πi+1
这里,令π0为起始状态,πi+1为终止状态。
在表示或分析HMM模型时,用方框表示各个状态,方框之间的连线表示状态转换。对于每个状态,详细地描述各个字符的释放概率,而对于状态之间的转换,也给出相应转换动作发生的概率,即状态转换概率。表示DNA序列的HMM如图1所示。
对生物序列而言,HMM的字符就是20个字母的氨基酸或4个字母的核苷酸。编码蛋白质的原始DNA序列,在生物的进化过程中会受到自然环境和各种因素的影响,使翻译出的蛋白质序列[4]经历突变、遗失或引入外援序列等变化,最后按不同的进化路径分化,形成多种功能相近的蛋白质。因此,可以把这些蛋白质看作由一个基本蛋白质序列经过插入、删除或替换了某些氨基酸残基而形成。这个过程可以用HMM来表示。一个训练好的模型可以代表有共同特征的蛋白质序列。HMM用于分析蛋白质序列的原理是分析蛋白质产生不同序列的概率,对于与模型相符合的序列,能以较大的概率产生。
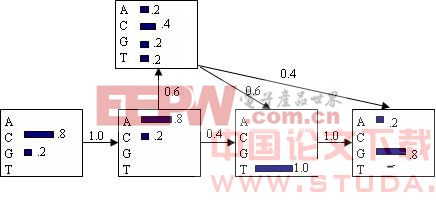
图1 隐马尔科夫模型
3 并行算法
对于给定一个隐马尔科夫模型M=(A,S,Q)和一个字符序列X(即基因序列),在M中寻找产生该序列的最优路径Π*,该路径从起始状态出发,结束于终止状态,在路径中的每一个状态都选择释放一个字符,使P(X|Π*)最大。这是基因识别中常用的一个方法,这里我们设计采用并行算法来求解HMM的最优路径问题。
给定一个字符序列X=(x1,x2,…,xL),以vk(i)代表序列前缀(x1,x2,…,xL)终止于k(k∈S,1≤i≤L)的最可能路径的概率。求解过程如下:
(1)初始化 vbegin(0)=1
k≠begin vk(0) = 0
(2)对于每个i=0,1,…,L-1及每个l∈S,按下式进行递归计算
vl(i+1) = el(xi+1)max{vk(i)fkl} k∈S
(3)最后,计算序列X终止于状态“end”最可能的路径概率,即P(X|Π*)的值
P(X|Π*) = max{vk(L)fk,end} k∈S
在实现中我们将隐马尔科夫模型使用一颗状态空间树及一个字符释放概率矩阵联合表示。如图2所示。
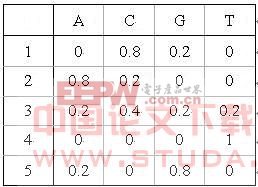
图2 HMM的联合表示
采用并行深度优先搜索技术,在每一个前向分支处启动一个新的进程,并行的计算多计算分支。在单CPU的情况下,算法的时间复杂度为O(L|S|2),在具有N个计算节点的情况下,算法的时间复杂度为O(L|S|2/N)。
在理想的情况下,并行算法在理论上的加速比与计算节点数成正比。在大型基因结构识别的问题域中,为实现并行计算而产生额外的启动、通信等时间与有效计算时间相比基本可以忽略,可近似达到理想加速比。
4 结束语
中国科学院院士张春霆指出生物信息学是生物学的核心和灵魂,数学与计算机技术则是它的基本工具。只有将并行计算研究和基因识别的理论研究有效联系起来,在研究蛋白质结构预测与分析的方法基础上,结合并行计算技术的特点,设计一系列的高效并行实现技术,实现高效、快速的基因识别,生物信息计算才能得到更快的发展。





评论